

�п�Ժ��������ˮϵ늽�Һ�о���ȡ���Mչ
�r�g:2021-11-15 19:29��Դ:�п�Ժ ����:�C�ψ��
�c��:
��
�����������ˮϵ����늳���鰲ȫ���Gɫ�͵ͳɱ��ܵ��Pע����������ˮϵ늽�Һ늻��W����խ��ˮϵ����늳�����ݔ��늉��ͣ������ܶȵͣ��Է�늸��Լ�ѭ�h������Ȇ��}���y���ƏV���á����ˌ��F���λˮϵ늽�Һ�������}���Water-in-salt늽�Һ�������ͨ�^����}��Ȍ��F�܄�-�x��������{�ƽ���ˮ��늻��W���Ժ͌��FSEI�x�ӌ��w�g��Ĥ����ˮϵ늽�Һ늻��W�����،���3V������(Science 350��6263��2015;JACS 139��51��2017)�����ǣ����S��늽�Һ�}��ȵIJ���������늽�Һճ�ȼ���������늌��ʿ����½�����Һ�D���ض�څ���Ҝأ����³����}���WIS늽�Һ�����W���ܺ͵͜�����׃�늽�Һ�ɱ����ߵȆ��}����ˣ�����ڌ��λ늽�Һ�Ќ��F�����Ą����W���ܺ͵͜����ܺ͵ͳɱ����ɞ�ˮϵ늽�Һ��ˮϵ����늳����R���P�I���g����
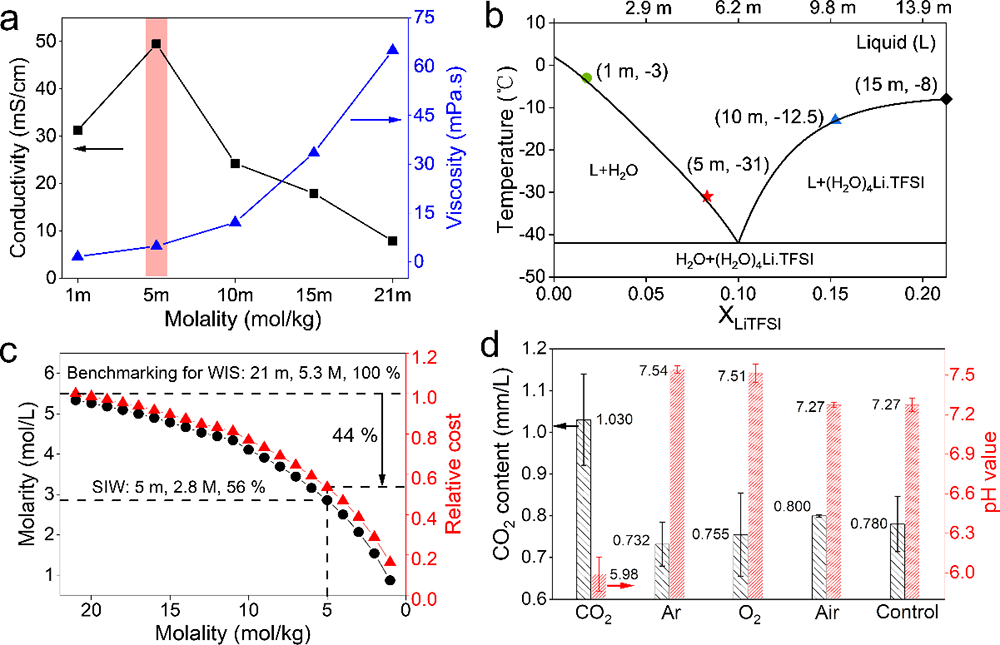
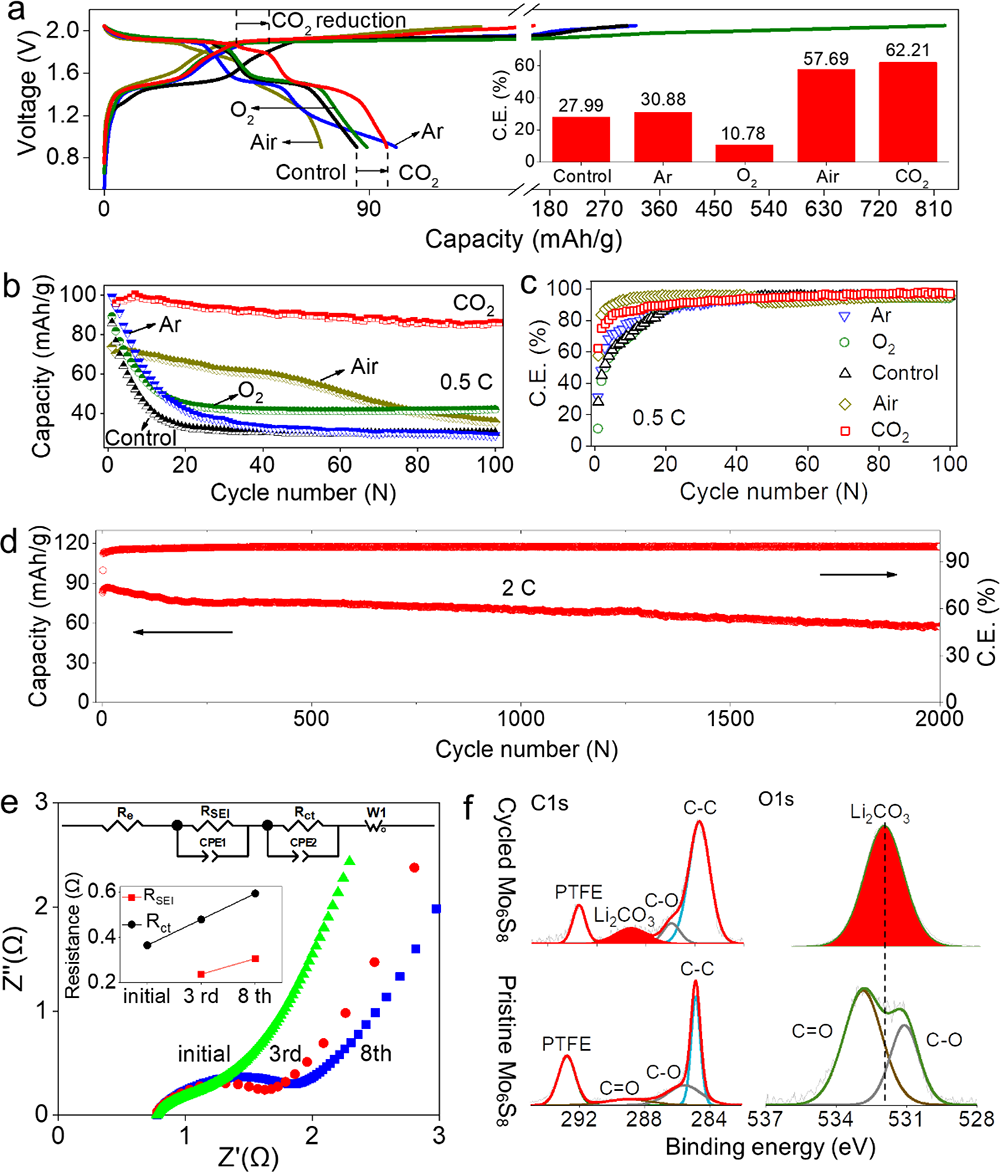
�������ڣ��Ї��ƌWԺ�����о���/�������ۑB��������о����đ����о������坍��Դ����Ҳ�ʿ���������о��T������ָ���£����о���ͬ�ܽ���w��SEIĤ�γɺͷ���Ӱ��^����(�D1��2)���l�FLiTFSIˮ��Һ�cCO2֮�g��������ď�����ã�ԓ���ÿ��Դ�ʹ�γɸ�CO2늽�Һ�wϵ�������S���늻��W�^������ؓ�O߀ԭ�γ�Li2CO3�����FSEIĤ�����W���o���Ķ����F늽�Һ늻��W���ڵ��،����Mһ���о��l�F���@�N������Ì��}�ĝ�Ⱦ�����ه�ԣ����ʬF������p��څ�ݣ��ڵ��}���5 m LiTFSI(Salt-in-Water��SIW)�r�(�D4)���c���y�����}���WIS(21 LiTFSI-H2O)늽�Һ��ȣ�CO2-SIW늽�Һ���������ஔ�Č�늻��W���ڣ������x��늌�����ǰ�ߵ�10����ճ�Ƚ�����һ������������-Һ�D׃�ض��½���50��(��-40��), �ɱ�������44 %(�D3)���@Щ�������W׃����δ���OӋ�ͳɱ��������ܡ���늉�ˮϵ늳��ṩ������g��
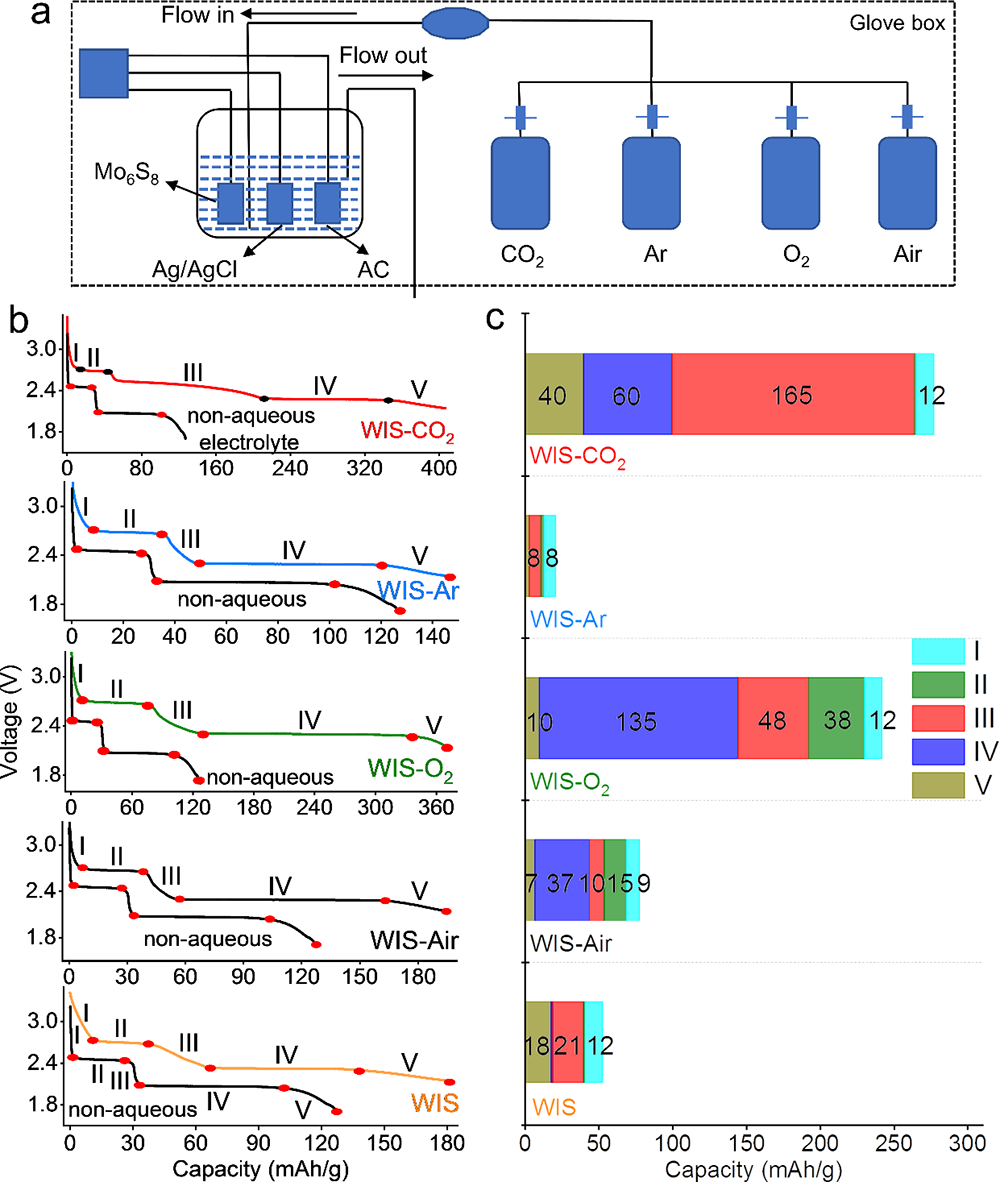
�����о��Mһ��������ø�CO2�ĵ��}���늽�Һ (5 m LiTFSI)(CO2-SIW)�팍�F��늉�ˮϵ��x��늳� (> 2 V)������ԓ�늽�Һ��2Vˮϵ��x��늳�(LiMn2O4-Mo6S8)���F��������늻��W���ܣ�����ѭ�h�^���Mo6S8ؓ�O�����γ�����Ч��SEI�g��Ĥ(�D5)�������ڵ��}���늌��ʸߡ�ճ�ȵ͡�Һ���c�ضȵ͵ȃ��ݣ�ˮϵ��x��ȫ늳����W���ܺ͵͜����ܵõ��˴����������Ҫ���F���ڳ�����d��늘O(334 um��40 mg/cm2)�Լ�-40 ��ؗ͜l���¿��Է�����Ч�������@��ԭ�г����}���WIS늽�Һ�o�����F��(�D6)��
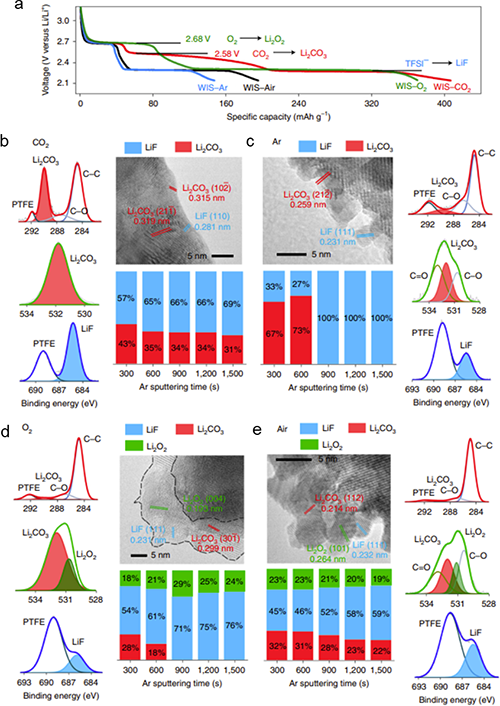
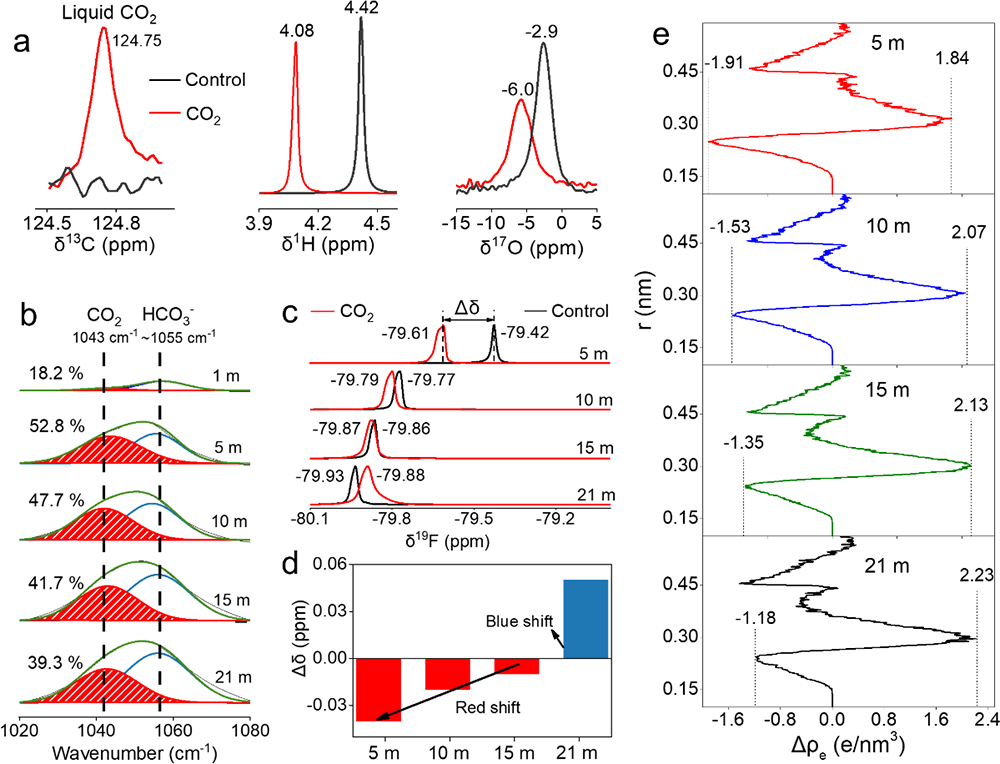
����ԓ�����о��ɹ����Y���£�(1)������늘O�b��ģ�M��늳حh�����ܽ���w��߀ԭ�����_�y����CO2��O2��߀ԭ�λ������߀ԭ�a�(CO2→Li2CO3��2.58 V vs. Li/Li+;(2)O2→Li2O2��2.68 V vs. Li/Li+)(�D1��2);(2)���������~׃�Q�t����V�ͺ˴Ź���ĽY�����Y��MDģ�M���C����CO2�cTFSI��x��֮�g���ڏ����������5 m̎�_�������5 m��CO2̎��һ�N�����ͷ����Ġ�B�����ױ����ߵ������������(�D4);(3)��CO2-SIW늽�Һ�У�ȫ늳�(LiMn2O4-Mo6S8)�ڵͱ��ʺ߱����¾��������õ�ѭ�h�����ԣ���0.5 C���M��100��ѭ�h����������90 %(��ʼ������95.94 mAh/g��100��ѭ�h��86.12 mAh/g)����2 C���M��2000��ѭ�h���������ֽ�70 %(��ʼ������83.12 mAh/g��100��ѭ�h��57.50 mAh/g)(�D5a-d);(4)��CO2-SIW늽�Һ��,ȫ늳�(LiMn2O4-Mo6S8)���F�����õĄ����W���ܡ��cWIS��ȣ�CO2-SIW���H���и��õı������ܣ���15 C�r���������ʸ��_70 %������ʹ�� 300 um���ϵij���Mo6S8늘O(���d���� 40 mg/cm2)�r����Ȼ���Ԍ��F90 mAh/g�ij�ʼ������50��֮��ı����ʸ��_88 %������-40 ��Ҳ����������, ��0.5 C�r���Ա���70 mAh/g��������90��֮��o���@˥�pڅ��(�D6);(5)�״��ڵ͝��늽��|��Һ���γ��˷�����SEI����CO2-SIW늽�Һ�У�ȫ늳�ѭ�hǰ��ѭ�h���Nyquist�D��XPS���V�Y���@ʾ��Li2CO3������SEI����ӣ�LiF������SEI�ăȌ�(�D5e-f)��
�������⣬���}���LiNO3늽�Һ��ˮϵ��x��늳����ѽ����Î�ʮ��(JES 142��6��1995)��ͬ�Ӵ����ܽ�O2��CO2���sδ�l�F��Li2O��Li2O2��Li2CO3���γɣ��f�����˸��}���֮�⣬��x�ӻ��WҲ���T���γ�SEIĤ���P�I���c�о��Y��һ�¡����ܽ�CO2�cTFSI��x���g���صď������ʹ��CO2������SIW늽�Һ����һ�N���������Ġ�B���ڣ��M����߀ԭ����SEI���o���á�
�����о������״ΰl�F��LiTFSI-CO2֮�g������������ã��@�N��������}-���w-ˮ�g���»��W�B�錍�F��늉�ˮϵ늳��ṩ��ȫ�µĽ����{���ֶΣ�ʹ���_�l���λˮϵ늽�Һ���چμ���ه�����}��ȁ팍�F���錒�λˮϵ늽�Һ���OӋ�_�l�ṩ����˼·��
�������P�о��ɹ���Aqueous interphase formed by CO2 brings electrolytes back to salt-in-water regime���}���l����Nature Chemistry�ϡ��о������õ�������Ȼ�ƌW�������������������ۑB��������о����ġ������坍��Դǰ���о����ġ������坍��Դ���Ϝyԇ�\���c�аlƽ�_�Լ��������ϻ��̸߾��ℓ�����ĵ�֧�֡�

(؟�ξ�������)
���˺���
늽�Һ




��؟�������ăH�������߂����^�c���c�Ї�늳��˟o�P����ԭ�����Լ�����������ֺ̓���δ�����W�C�����������Լ�����ȫ�����߲��փ��ݡ����ֵ��挍�ԡ������ԡ����r�Ա�վ�����κα��C����Z��Ո�x�߃H����������Ո���кˌ����P���ݡ�
�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@cbcu.com.cn
�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@cbcu.com.cn
����ϲ�g
-
�늳�늽�Һ���ӽY���U��
2021-09-16 12:24 -
�����U���ψF��OӋ����늽�Һ��������Q�����늳�ѭ�h�����y�}
2021-06-04 10:10 -
�K�ݼ{�����OӋ�������x��Һ�w���늳ذ�ȫ늽�Һ
2021-04-09 11:27 -
�о��ˆT�аl����ꎘO��늽�Һ ʹ�߹����V늳سɞ����
2020-12-04 09:54 -
���Ȼ�÷¡��W�����˹����ܷ����_�l늽�Һ �ӿ�늳��²���
2020-12-04 08:53 -
�������MԺ�аl�����ڸ�늉��ߝ��늽�Һ��⛻��pʯī늳�
2020-09-28 17:09 -
���}���늽�Һ�ٽ��c�x��늳سɱ�
2020-09-17 09:53 -
���������x��Һ�wȡ�����y�ЙC늽�Һ �аl����ȫ/���־Ã����O��
2020-09-08 08:58 -
˹̹���аl����늽�Һ��䇽���늳� �ɜp�p늄���܇����/���L�m��
2020-07-23 11:25 -
˹̹����W�l������늳�늽�Һ�������늄���܇��������
2020-06-28 09:59
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

���}


���P��
-
�늳�늽�Һ���ӽY���U��
2021-09-16 12:24 -
�����U���ψF��OӋ����늽�Һ��������Q�����늳�ѭ�h�����y�}
2021-06-04 10:10 -
�K�ݼ{�����OӋ�������x��Һ�w���늳ذ�ȫ늽�Һ
2021-04-09 11:27 -
�о��ˆT�аl����ꎘO��늽�Һ ʹ�߹����V늳سɞ����
2020-12-04 09:54 -
���Ȼ�÷¡��W�����˹����ܷ����_�l늽�Һ �ӿ�늳��²���
2020-12-04 08:53 -
�������MԺ�аl�����ڸ�늉��ߝ��늽�Һ��⛻��pʯī늳�
2020-09-28 17:09 -
���}���늽�Һ�ٽ��c�x��늳سɱ�
2020-09-17 09:53 -
���������x��Һ�wȡ�����y�ЙC늽�Һ �аl����ȫ/���־Ã����O��
2020-09-08 08:58
�����c
-
늳�ԭ���σr���A�گ��q
2021-11-03 09:57 -
���Y��������늳�֮�����S��20�ꡰ���ӡ��ı��K����
2021-10-16 12:29 -
�����f���Ƴ�����Һ��늳ؼ��g ������I���F̼�к�Ŀ��
2021-11-01 21:42 -
����䇡����֮�£�늳؏S���١�ٍ�X��
2021-10-16 11:52 -
���������@���늳ػ�����I������
2021-10-20 11:36 -
�������F�늳�2021-2025���Ј������������L�ʵĜy��
2021-11-04 10:23 -
�|������ٺ���������늳��Ŀ ��ȶ��δ��ֹPͶ�Y
2021-10-29 10:56 -
�A��������AEM����-50��C�����\�е�ȫ�����x��늳أ�
2021-10-18 11:11

©2017 ������� 늳��� �A����̩�Ƽ�������������˾ ���k Power by DedeCms
�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
 ��I��̖
��I��̖ �Ź���̖
�Ź���̖